| Examples
Molecule in cartesian coordinates
The following file is that of water molecule. Cartesian coordinates have been
used to specify the geometry.
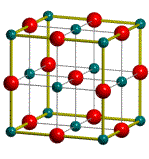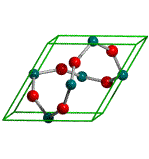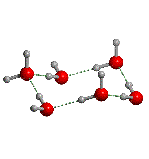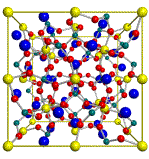
TITLE
Ice D2O Peterson et al Acta Cryst. 10, 70
(1957)
CELL
1 1 1 90 90 90
COORD
8
0.0000000000 0.0000000000 0.0000000000
1 0.3368178282 0.9390651487
0.0000000000
1 -0.9976429393
0.0000000000 0.0000000000
-1 0 0
0
GROUP
P1
Molecule in Z-matrix coordinates
The next example is also water but specified in Z-matrix internal
coordinates.
TITLE
water
CELL
1 1 1 90 90
90
MATZ
1 0.0
0 0.0 0 0.0
0 0 0 0
8
0.97 0 0.0
0 0.0 0 1 0
0
1 0.97 0
104.5 0 0.0 0 2
1 0
0 0
0 0 0
0 0 0 0 0
Molecular crystal in fractional coordinates
Here is shown the mol file for the benzene crystal as determined by X-ray
diffraction. Please beware that only the asymmetric unit of the benzene crystal
is given in the original MOL file:
TITLE
Benzene
CELL
7.44000 9.55000 6.92000 90.00000 90.00000
90.00000
COORD
6 -0.05690 0.13870 -0.00540
6 -0.13350 0.04600 0.12640
6 -0.07740 -0.09250 0.12950
1 -0.09760 0.24470 -0.01770
1 -0.24090 0.07940 0.22180
1 -0.13710 -0.16310 0.23120
0 0 0 0
Once MOLDRAW read the structure for the first time the input file
becomes:
TITLE
Benzene
CELL
7.44000 9.55000 6.92000 90.00000 90.00000
90.00000
COORD
6 -0.05690 0.13870 -0.00540
6 -0.13350 0.04600 0.12640
6 -0.07740 -0.09250 0.12950
1 -0.09760 0.24470 -0.01770
1 -0.24090 0.07940 0.22180
1 -0.13710 -0.16310 0.23120
0 0 0 0
BARICE
-.704044678137831 .310460506695342 .610824255317637
CONNB
5
1 2 1 4 2 3 2 5 3 6
BONDS
2 3 2 1 1 1
FRAG
1
1 1 1 1 1 1
The meaning of the keyword added by MOLDRAW is reported here.
The structures is displayed as:
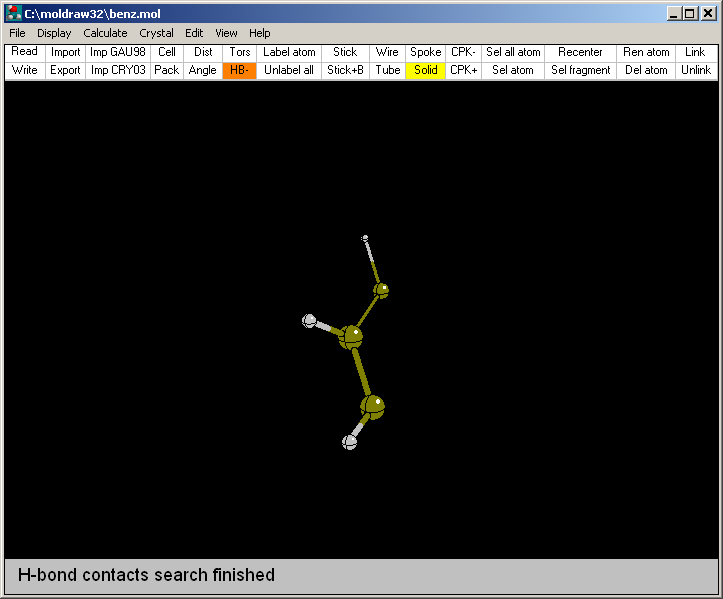
i.e. only half benzene molecule is shown, which is the
asymettric unit resulting from the X-ray crystallographic determination. To see
a larger portion of the benzene molecular crystal the space group information should be provided.
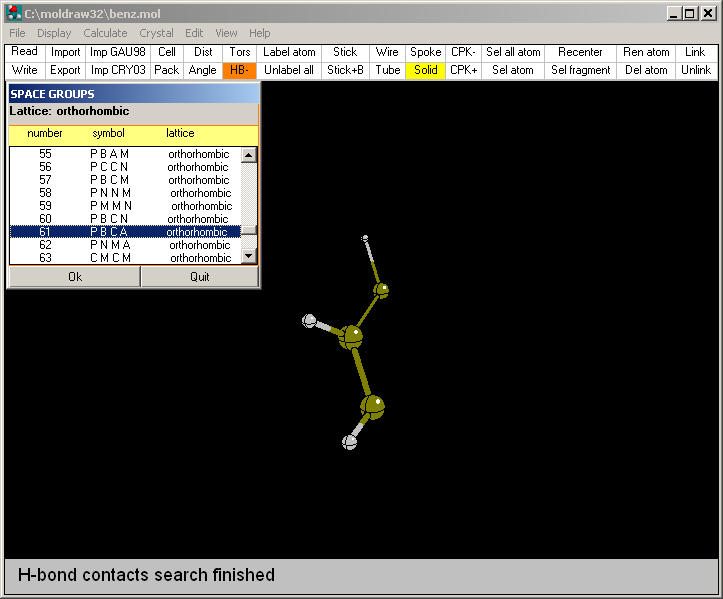
To achieve that open the (Crystal--->Select a space
group...) menu. Automatically MOLDRAW will provide only the space
groups which are compatible with the lattice symmetry (in this case the benzene
lattice is orthorombic and the space
group is P B C A).
The corresponding MOL file is then updated with the space group and symmetry
records for future use.
TITLE
Benzene
CELL
7.44000 9.55000 6.92000 90.00000 90.00000
90.00000
COORD
6 -0.05690 0.13870 -0.00540
6 -0.13350 0.04600 0.12640
6 -0.07740 -0.09250 0.12950
1 -0.09760 0.24470 -0.01770
1 -0.24090 0.07940 0.22180
1 -0.13710 -0.16310 0.23120
0 0 0 0
BARICE
-.704044678137831 .310460506695342 .610824255317637
CONNB
5
1 2 1 4 2 3 2 5 3 6
BONDS
2 3 2 1 1 1
FRAG
1
1 1 1 1 1 1
GROUP
P B C A
SYMNUM
0 0 0 1 0 0 0 1 0 0 0 1
0.5 0 0.5 -1 0 0 0 -1 0 0 0 1
0.5 0.5 0 1 0 0 0 -1 0 0 0 -1
0 0.5 0.5 -1 0 0 0 1 0 0 0 -1
0 0 0 -1 0 0 0 -1 0 0 0 -1
0.5 0 0.5 1 0 0 0 1 0 0 0 -1
0.5 0.5 0 -1 0 0 0 1 0 0 0 1
0 0.5 0.5 1 0 0 0 -1 0 0 0 1
-1. -1. -1. 0 0 0 0 0 0 0 0 0
It is now possible to build a piece of crystal by the Pack label on the toolbar:
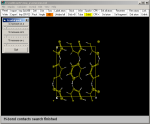
Now a complete benzene molecule is surrounded by its symmetry related
replicas.
You are here: Home-Navigate-Main features-File format-Examples
Previous Topic: Program keywords Next Topic: Import from...
|