MOLDRAW allows to add atoms to a preexistent structure in a rather
flexible way. Because no force filed is provided the geometry should be arranged
by the user.
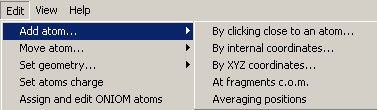
The (Edit-->Add atoms...) menu provides all the
needed options:
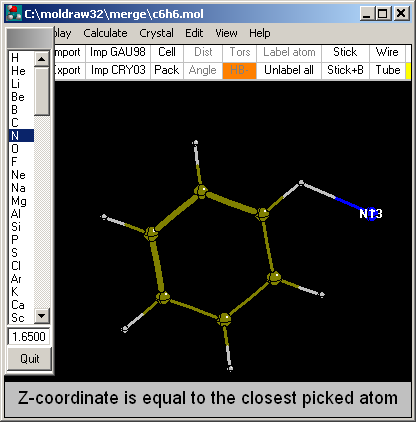
Option: Clicking close to an atom...
This is the quickest way to add an atom and can be
surprinsigly accurate as well. It is enough to position the mouse close to
the atom to which it will be bonded (in the picture it was close to the
H). MOLDRAW will automatically add the atom (in this case a N) exactly
positioned where the mouse cursor has been set, but for the distance which
is set in the toolbar (in this case it was decided to be 1.65). The added
atom always has Z=0.0. The process can be repeated as many times as
needed. To add atoms with a Z different from 0 rotate the
structure
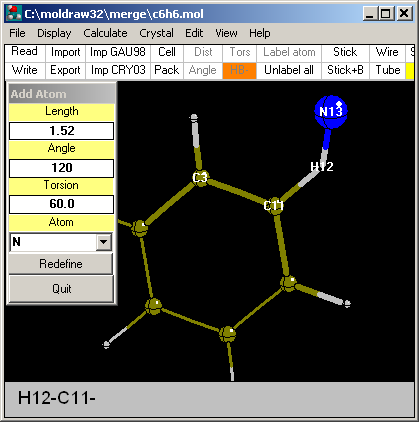
Option: By Internal coordinates...
This is the most accurate way to add atoms. It is based on
the specification of bond length, bond angle and bond torsion angle with
respect to three preexistent atoms which are clicked in sequence. In the
case shown in the picture, a N atom is added to H12 (bond lenght=1.52 A),
making an angle of 120 degrees with C11 and a torsion angle of 60 degrees
with the C3 atom (torsion angle N13-H12-C11-C3). All geometrical
parameters can be modified and the Redefine button set them on the fly. Atom
type is chosen via the dragdrop menu.
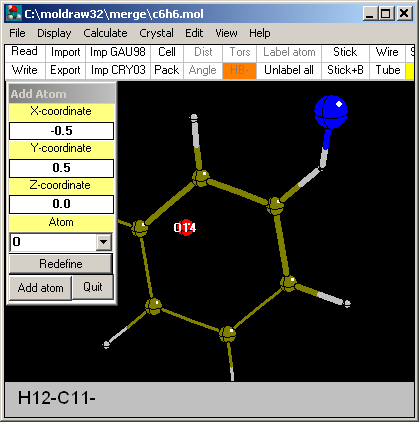
Option: By XYZ coordinates...
This is the most straightforward way to add atoms. It is
based on absolute values of the atomic coordinates. Considering that a
given structure is always centered in X=0.0, Y=0.0, Z=0.0 it becomes
relatively easy to locate different points. This method is very useful for
extremely symmetric structures in which the atoms are positioned in
special positions. In the case shown in the picture, an O atom is added at
X=-0.5, Y=0.5 and Z=0.0. Fine tuning of the coordinates is possible via
the Redefine button. Atom type
is chosen via the dragdrop menu.
Option: At fragment c.o.m.
In this case a dummy atoms (LP type) is automatically added at the
center of masses of the structure present in memory.
Option: Averiging positions
In this case a dummy atoms (LP type) is added at the center of
masses of a selected number of atoms. The user can repeat the operation for as
many times is needed.